
티스토리 뷰
gplots을 이용해 heatmap을 자주 그리게 된다. 하지만 cluster 정보를 자세히 들여다 보고싶을때 어떤 구간이 몇번째 클러스터로 지정된것인지 확인하는데 복잡한부분이 있어서 오늘 그 점을 어떻게 해결했는지 이야기하려고 한다.
Getting library
library(gplots)
library(dplyr)Example data set
data <- matrix(rnorm(500), 20, 5, dimnames=list(paste("gene", 1:20, sep=""), paste("S", 1:5, sep="")))
S1 S2 S3 S4 S5
gene1 1.7288141 -1.21826069 0.1223872 0.887119088 2.1037146
gene2 -1.5198759 -0.54651642 0.7109011 -1.640756604 0.4192511
gene3 0.9725800 -0.04533105 0.4879282 -1.117202159 -0.1103785
gene4 0.3650084 0.04452878 2.5910805 -1.087627097 0.9907894
gene5 -1.4630352 -0.07544341 -0.2952500 0.179749634 0.1828693
gene6 -1.8396656 -2.38672198 0.3261195 0.002694406 0.2323069이 데이터를 이용해서 기본heatmap을 그려보면 다음과 같은 결과가 나온다.
library(gplots)
heatmap.2(data)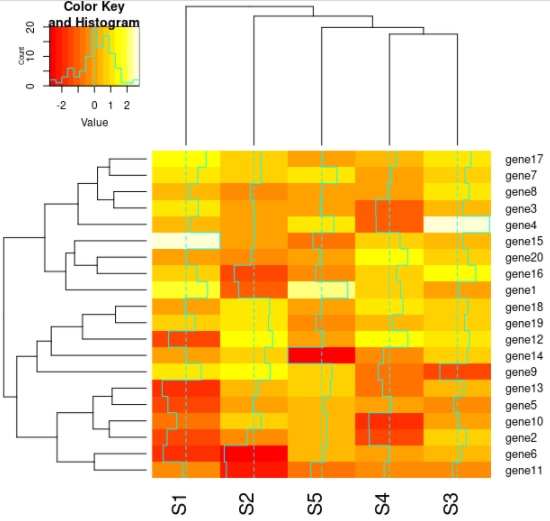
Clustering with hclust
이제 row, column을 hclust를 이용해서 clustering을 진행한다.
## Row- and column-wise clustering
hr <- hclust(as.dist(1-cor(t(data), method="pearson")), method="complete")
hc <- hclust(as.dist(1-cor(data, method="spearman")), method="complete")
hr결과를 예시로 확인해보면 hight, order, labels등의 정보가 들어있다.
> head(hr)
$merge
[,1] [,2]
[1,] -14 -19
[2,] -11 -16
[3,] -12 -18
[4,] -15 -17
[5,] -2 -4
[6,] -5 -13
[7,] -8 2
[8,] -10 5
[9,] -6 -20
[10,] -3 -7
[11,] 1 4
[12,] 3 6
[13,] -9 10
[14,] 7 9
[15,] -1 13
[16,] 8 12
[17,] 14 16
[18,] 11 15
[19,] 17 18
$height
[1] 0.06951097 0.08027382 0.09776064 0.10953254 0.15069633 0.19519260 0.38423985
[8] 0.40287137 0.40606194 0.41298017 0.61689002 0.63439827 0.66761664 0.74465629
[15] 0.92154820 1.22156155 1.73439354 1.77268196 1.95526914
$order
[1] 8 11 16 6 20 10 2 4 12 18 5 13 14 19 15 17 1 9 3 7
$labels
[1] "gene1" "gene2" "gene3" "gene4" "gene5" "gene6" "gene7" "gene8" "gene9"
[10] "gene10" "gene11" "gene12" "gene13" "gene14" "gene15" "gene16" "gene17" "gene18"
[19] "gene19" "gene20"
$method
[1] "complete"
$call
hclust(d = as.dist(1 - cor(t(data), method = "pearson")), method = "complete")
이결과를 이용해서 heatmap을 그려보면, order순서와 heatmap에서의 gene 순서의 관계를 볼 수 있다.
heatmap.2(data, Rowv=as.dendrogram(hr), Colv=as.dendrogram(hc), scale="row", density.info="none", trace="none")

Tree Cutting with cutree
tree cutting에는 두가지 방법이 있다.
- k : the number of clusters the tree should be cut into
- h : a hight where the tree should be cut
여기서는 h 방법을 이용했다.
# cluster method-1 : cutting by distance
cutoff <-0.9
cluster <- cutree(hr, h=cutoff)
# cluster method-2 : cutting by number of subcluster
# cutoff <- 5
# cluster <- cutree(hr, k=cutoff)cluster 결과를 보면 각 유전자가 몇번의 클러스터에 지정되었는지 알 수 있다.
> cluster
gene1 gene2 gene3 gene4 gene5 gene6 gene7 gene8 gene9 gene10
1 2 3 2 4 5 3 5 3 2
gene11 gene12 gene13 gene14 gene15 gene16 gene17 gene18 gene19 gene20
5 4 4 6 6 5 6 4 6 5 mycolhc <- rainbow(max(cluster), start=0.1, end=0.9)
mycolhc <- mycolhc[as.vector(cluster)]
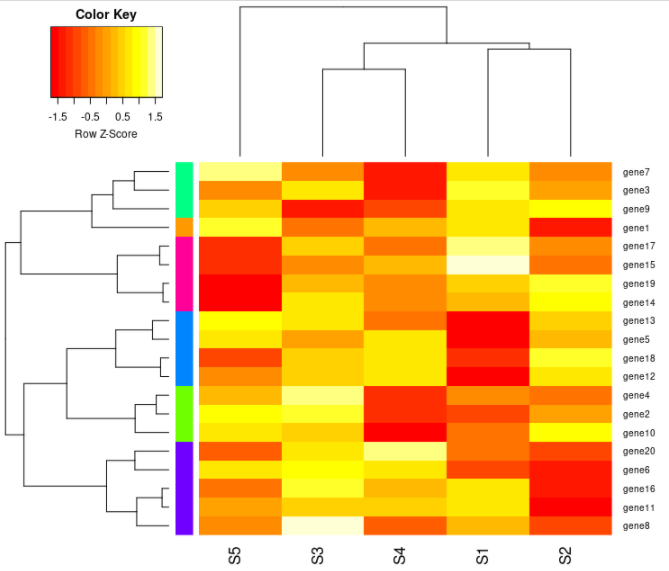
heatmap.2(data, Rowv=as.dendrogram(hr), Colv=as.dendrogram(hc), scale="row", density.info="none", trace="none", RowSideColors=mycolhc)
이결과를 heatmap으로 그려보면 다음과 같다. 하지만 여기서 주의할 것은 왼쪽의 클러스터가 위부터 순서대로 진행되지 않는다는 점이다. 그 이유는 아마도 gene 순서대로 번호가 부여되어서 그런것 같다. 그래서 클러스터 번호를 순서에 맞춰 새로 지정해주는 과정이 필요했다.
Re-ordering Cluster Numbers
data_order <- data.frame(label = hr$labels[hr$order], sample = hr$order, order = seq(length(hr$order)))
data_order <- data_order[order(data_order$sample),]
data_order <- data.frame(data_order,cluster)
data_order <- data_order[rev(order(data_order$order)),]
mycolhc <- rainbow(max(cluster), start=0.01, end=1)
check <- data_order$cluster[1]
data_order$cluster_new <- 0
data_order$color <- ""
new_n <- 1
for (i in seq(nrow(data_order))){
data_order$cluster[i]==check
if (data_order$cluster[i]!=check){
new_n = new_n+ 1
check <- data_order$cluster[i]
}
data_order$cluster_new[i] <- new_n
data_order$color[i] <- mycolhc[new_n]
}> data_order
label sample order cluster cluster_new
20 gene7 7 20 3 1
19 gene3 3 19 3 1
18 gene9 9 18 3 1
17 gene1 1 17 1 2
16 gene17 17 16 6 3
15 gene15 15 15 6 3
14 gene19 19 14 6 3
13 gene14 14 13 6 3
12 gene13 13 12 4 4
11 gene5 5 11 4 4
10 gene18 18 10 4 4
9 gene12 12 9 4 4
8 gene4 4 8 2 5
7 gene2 2 7 2 5
6 gene10 10 6 2 5
5 gene20 20 5 5 6
4 gene6 6 4 5 6
3 gene16 16 3 5 6
2 gene11 11 2 5 6
1 gene8 8 1 5 6이 과정을 거치면 새로운 클러스터 번호를 부여받을 수 있고, 어떤 유전자가 몇번째 클러스터에 속하는지 쉽게 파악 할 수 있다.
참고
'R' 카테고리의 다른 글
| R | Rstudio를 이용하지 않고 현재 파일이 있는 디렉토리 위치 불러오기 (0) | 2021.05.06 |
|---|---|
| R | Rstudio에서 현재 파일이 있는 디렉토리 위치 불러오기 (0) | 2021.01.07 |
| R | COVID-19 데이터로 무엇을 할 수 있을까 | 4. 확진자 증가 추세 추적 그래프 (0) | 2020.05.07 |
| R | ceiling()를 이용한 올림, floor()를 이용한 내림 (0) | 2020.05.06 |
| R | round()를 이용한 반올림 (0) | 2020.05.06 |
공지사항
최근에 올라온 글
최근에 달린 댓글
- Total
- Today
- Yesterday
링크
TAG
- data
- 2진수
- plot
- SEQ
- 엑셀
- covid
- Python
- RStudio
- hist
- format
- geom_line
- Excel
- Coding
- Command
- Cast
- geom_bar
- 숫자
- r
- coronavirus
- visualizing
- for loop
- Order
- Visualization
- Heatmap
- 데이터
- 팟빵
- 파이썬
- 코로나바이러스
- comma
- BIOINFORMATICS
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||
| 5 | 6 | 7 | 8 | 9 | 10 | 11 |
| 12 | 13 | 14 | 15 | 16 | 17 | 18 |
| 19 | 20 | 21 | 22 | 23 | 24 | 25 |
| 26 | 27 | 28 | 29 | 30 | 31 |
글 보관함